In this blog post, I’d like to quickly go over some very basic techniques in molecular biology. I’m going to stick strictly to the big ideas; these ideas should be enough to get you reading papers, but certainly not enough to be performing these procedures in a lab.
Plasmid transformation
Plasmid transformation is a mechanism by which we can introduce genes of interest into bacterial cells. In order to do this, we isolate another plasmid, insert the gene we want, and then put the plasmid back into the bacterial cells.
We can isolate the plasmid via various biochemical means, and plasmids are small enough such that we can isolate whole plasmids. (We have difficult isolating whole pieces of longer DNA, which tends to break up into fragments.) Once we have isolated the plasmid, we can apply restriction enzymes (such as EcoRI) for the specific sequences that we want to the plasmid and the gene of interest. The restriction enzymes cleave the plasmid and our gene at cut sites; often, this leaves stick ends (ends of DNA with a single-stranded strand between two and four bases long). These sticky ends readily bond back together with other matching DNA pieces; in this manner, we can join the gene of interest into our plasmid or cut other pieces out of the plasmid. After adding the restriction enzyme, we add DNA ligase to ligate the plasmid back together (to reform the covalent bonds that hold the DNA together).
Once we have our ligated plasmid, we would like to insert it into bacterial cells. Some bacteria are naturally competent, and will simply accept plasmids from their environment. However, many other bacteria (such as E. Coli), are not naturally competent, and we have to induce competence in order to get them to accept the plasmid. There are electrical methods to induce competence, which apply a field and essentially break the bacteria open in order to accept the plasmid; although most of the bacteria die, some survive and form colonies. There are also chemical methods to induce competence, such as adding calcium chloride to E. Coli, but their mechanism of action is more complicated.
Finally, once we have inserted our plasmid into bacterial cells, we can grow them back into colonies. If we need to, we can apply selection for recombinant plasmids, which is discussed later in this technique primer.
Restriction digestion
A restriction enzyme is a protein which cleaves DNA at specific sequences. The resulting break in the DNA can be used to insert other pieces of DNA into that location, after which DNA ligase can be used to join the DNA back together.
The sequences at which restriction enzymes operate are usually the same from 5’ to 3’ (they are DNA palindromes). The reason for this is that restriction enzymes are usually dimers where each monomer in the dimer operates on a single strand. If the sequence is palindromic, then both monomers will bind to the same location on the strand.
Restriction enzymes are specific to sequence and also the type of cut they make. They can create blunt ends in the cut (where the cut is in the middle of the sequence), resulting in entirely double stranded DNA, all the way up until the break. They can also create what is known as “sticky ends”. A sticky end is a piece of DNA which at end of one strand has a small single-stranded portion, which is created because the restriction enzyme cuts the sequence off-center. This single-stranded portion can be between two and four bases long, and matches the single stranded portion on the other half of the break. Since they match, having them join together is energetically favorable (since it forms hydrogen bonds between the single-stranded portions), so it is more likely to happen; this is why these are referred to as “sticky” ends.
When the DNA joins back together with hydrogen bonds, there is no reason why it should join back in the exact same configuration as before. Because of this, we can cleave DNA and insert other pieces of DNA (with matching sticky ends) into that region. After that, we can use DNA ligase to covalently bond the pieces of DNA back together.
Electrophoresis
Electrophoresis is a process by which we can separate DNA fragments based on their molecular weight, and visually inspect what different sizes exist, as well as measure the sizes of the DNA fragments.
In order to do DNA electrophoresis, we begin by preparing the DNA fragments we’re interested in. In some cases, these come from restriction enzymes being applied to larger fragments, but that is in no way required or a characteristic of electrophoresis. We then create an agar gel which is put into a plate; this plate must have two electrodes on the sides.
Once the plate is prepared, we place DNA in wells in the agar gel on the side of the negative electrode. An electric field is applied between the two electrodes, and due to the negative charge on the phosphates, the DNA migrates towards the positive electrode. The electric field is held on for a while and then turned off. Smaller fragments will encounter less resistance from the gel, and will thus travel faster, while larger fragments which have a harder time getting through pores in the gel will travel slower. The distance traveled is related more or less logarithmically to the size of the molecule, because smaller pieces experience exponentially less resistance than larger ones.
Note that if you have fragments of similar size, it may be impossible to discriminate between them, because they will be placed next to each other. However, you can increase the time period over which the gel is run to reduce this.
In order to visualize the results, we add ethidium to the gel (or afterwards). Ethidium is a dye which binds to the DNA and sits inside the DNA helix. As the DNA migrates, it collects ethidium and thus has it in much greater concentrations than the surrounding gel. Ethidium glows under ultraviolet light, thus allowing us to see where the bands of DNA are.
For large pieces of DNA, gel electrophoresis may be ineffective, because the large pieces all will move at the same speed. In order to get around this, we can use pulsed electrophoresis. In this case, there are more electrodes on the edges, and the field points always towards the positive end of the plate but diagonally up or down; the field is pulsed in these two directions. The directions average out to a straight line, but due to the fact that the field changes rapidly, the DNA must reorient itself. Larger fragments take longer to reorient themselves, and thus this technique may be used to separate large DNA fragments that would otherwise travel together.
Given DNA marker fragments of known size, we can use electrophoresis to compute the size of the fragments. We can also use this technique for the purpose of sequencing DNA (Sanger sequencing).
Ligation
DNA Ligase is an enzyme which is responsible for covalently joining together pieces of DNA or nucleotides. In in vivo DNA replication, ligase fills in the nicks (un-bound DNA pieces) left after lagging strand replication. (Recall that the RNA primers are degraded after which DNA polymerase adds bases to fill in the gap left by the primer, but it cannot covalently join the new bases to the bases that were there after the primer; this is what is done by DNA ligase.)
We can use DNA ligase in combination with restriction enzymes to cut up DNA and then put it back together. For instance, we can use this to create recombinant plasmids.
Ligase works better with sticky ends left by restriction enzymes, but will work fine with blunt ends as well, although higher concentrations will be required.
Selection for recombinant plasmids
When creating recombinant plasmids and transforming bacteria with them, we may end up with bacteria with both the recombinant and original version of the plasmid. However, we may want to select only for the recombinant plasmid. In some cases, this is natural, and we can do so by just selecting for the property that the recombinant plasmid gives us.
Often, however, we will need to use more advanced techniques to select for the recombinant plasmid. In order to do this we can add in a second gene of interest, which we can then use to select for our plasmid. For instance, we can add a gene for a specific antibiotic resistance; then, we can grow our transformed bacteria on that antibiotic. The bacteria that have the original plasmid will die, while our transformed bacterial cells (which have that extra gene) will be selected for.
We can also do similar things with other genes. For instance, we could insert a fluorescent gene such as GFP. Once the colonies grow, we can examine them visually and select the ones that exhibit the fluorescence we want. This way, we can select only the colonies that have our plasmid.
Sanger Sequencing
Sanger Sequencing is a technique for obtaining the sequence of bases (As, Cs, Gs, and Ts) that compose some piece of DNA. It is one of the first methods developed, and, with minor improvements, is still in use today. Although it has much lower throughput than modern methods, it can sequence significantly larger contiguous sequences (as opposed to shotgun sequencing methods).
We begin by setting up a modified DNA synthesis reaction. In addition to adding template strand and primer (which determines what we are sequencing), we add sufficient amounts of deoxyribonucleotides (dNTPs). However, we also add a small proportion of dideoxyribonucleotides (ddNTPs) to the mixture; the ratio of ddNTPs to dNTPs is very small, on the order of one part to a hundred.


We then let synthesis proceed as we do in a normal in vitro DNA synthesis reaction. Note that we actually run four separate reactions, one for each type of ddNTP (ddATP, ddCTP, ddGTP, ddTTP). When we let this synthesis run, at each base which requires a given dNTP, there is some small probability of incorporating a ddNTP.
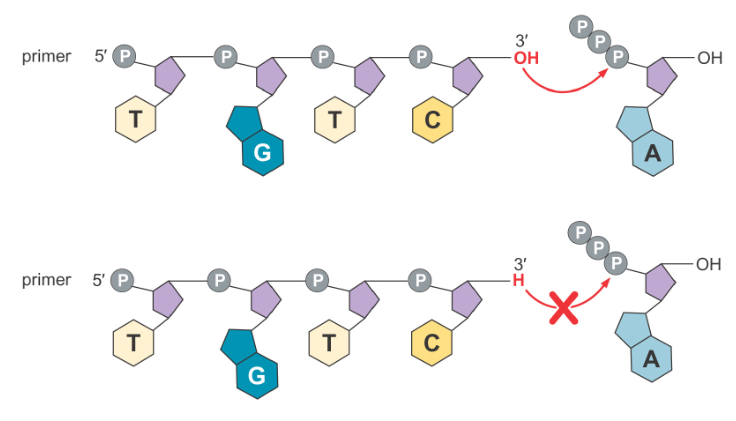
However, every time a ddNTP is incorporated, synthesis halts, because no new bases may be attached to the 3’ end of the resulting molecule. As a result, the synthesis will produce strands of varying length, with a break at every point where the present ddNTP could be incorporated. Although it is possible that a ddNTP is never incorporated at a given site, it is highly unlikely (statistically speaking) because the reaction is being run with many strands of DNA at once, not just a single one.
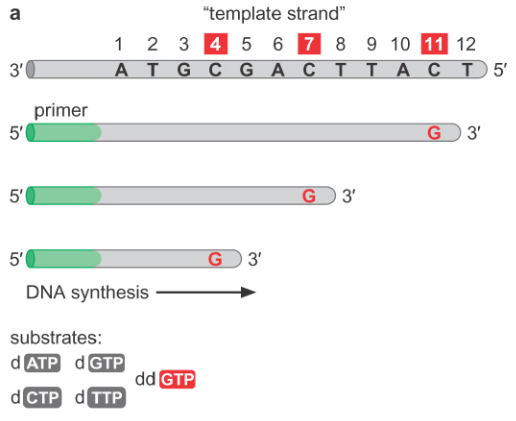
Since we have strands of different lengths, we can use gel electrophoresis to visually separate the strands. Note that we need to use a polyacrylamide gel instead of an agarose gel, because polyacrylamide gels have a much greater resolution than agarose gel, and can be used to resolve the length difference corresponding to a single base. When we put all of the four reactions we ran into four lanes of the same gel, we can view all of the different sizes produced. In order to make sure we get only the synthesized single strands and not the double stranded DNA, we run the gel under denaturing conditions, which correspond to high temperatures and the addition of chemicals to prevent hydrogen bonding.
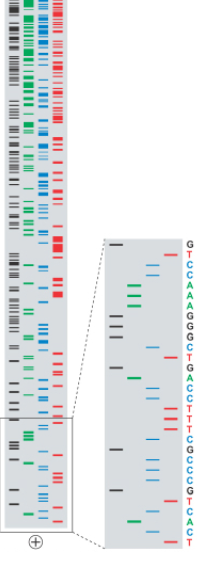
Finally, we can read off the sequence corresponding to the DNA by starting from the beginning of the gel and looking at the dark bands in the gel. Each position should have exactly one band in the four gels, which tells you which nucleotide is at that position. This method allows you to sequence long fragments, with length mostly limited by the fact that larger strands require larger gels and it becomes hard to distinguish different length sequences the larger the sequences are.
There are a number of modern improvements to this method which make it practical. For instance, instead of using four lanes of an electrophoresis gel and a radioactive phosphate marker, a single capillary tube is used with fluorescent markers of different wavelengths for each nucleotide type. A laser is placed over the capillary tube which measures the color under it at any given instant. Then, the dimension of interest is no longer length of the gel but instead time, producing a series of peaks for every wavelength.
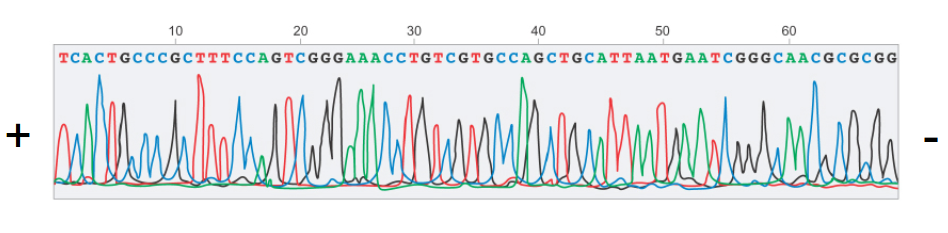
Using signal processing techniques, we can detect the peaks in the signal seen above, and extract the sequence of nucleotides, since each peak corresponds to a single nucleotide. Using current techniques, Sanger sequencing can be made to work efficiently and cheaply, sequencing DNA pieces of 300–1000 base pairs.
Southern and Northern Blotting
Southern and Northern blotting are techniques to visualize relative amounts of particular DNA or RNA sequences in particular samples of each. The original inventor (after whom the technique was named), Edwin Southern, performed this technique on DNA; later, it was used on RNA as well, and was dubbed Northern blotting when applied to RNA. For the remainder of this explanation, I’ll refer to the sequences being targeted as DNA, which is the case for Southern blotting, but remember that Northern blotting is simply the same thing with an RNA target sequence.
As usual, we begin by isolating the DNA (or RNA, in the Northern variant) that we wish to analyze. We put this DNA onto some electrostatically interacting surface, such as nitrocellulose or nylon. The DNA backbone binds (electrostatically) to the surface; note that the bases are still exposed for hydrogen bonding when the DNA is bound. The sheet of nitrocellulose or nylon is then either heated or exposed to ultraviolet light in order to permanently attach the DNA to its surface. At that point, extra DNA is added in order to cover any parts of the sheet that were not covered by the original DNA. This prevents the probe from binding to the sheet itself. Also, chemicals are added to reduce non-specific binding of the probe. Finally, a probe (usually RNA) is added. This probe has radioactivally marked elements or fluorophores attached to it, so that when it binds to its complementary DNA, it is visible. After hybridization of the probe, excess probe is washed away, and the sheet is imaged.

The result is an image which has varying brightness — dark regions correspond to high concentrations of the probe, while light regions correspond to low or nonexistent concentrations of the probe. Since the probe is designed to bind to a very specific DNA sequence, this allows us to measure the relative concentrations of DNA sequences in different samples.
In addition to using hand picked samples (as in a dot blot), we can combine gel electrophoresis with blotting in order to measure relative concentrations of DNA sequences at the same time as measuring the lengths of those sequences.
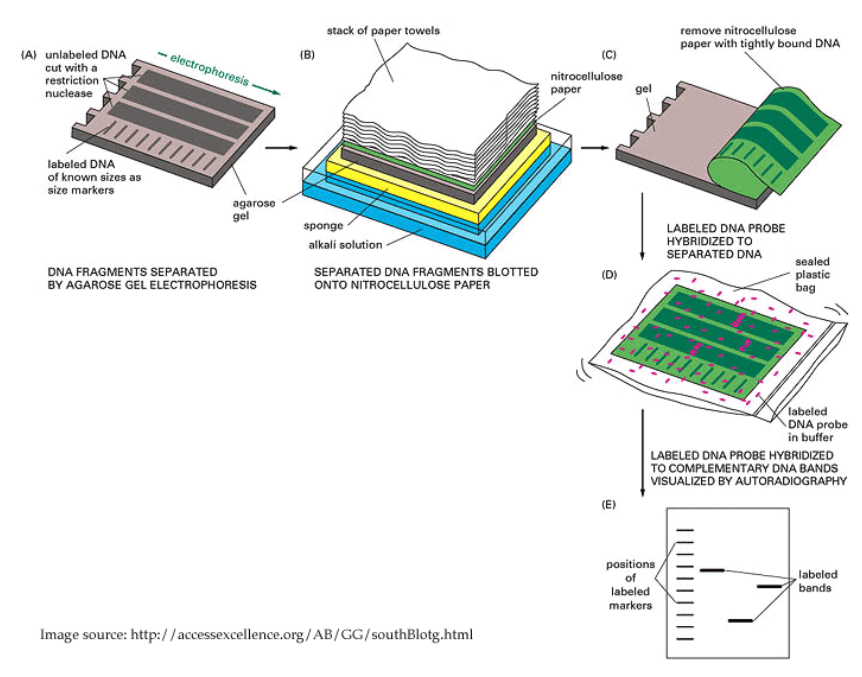
In this case, the gel electrophoresis reaction is run, and the membrane is placed between the gel and a stack of paper towels. The capillary action due to the paper towels pulls the DNA solution upwards, and the DNA gets deposited onto the membrane in the location right above where it was in the gel.
We get a result like what you see above — namely, dark bands corresponding to high concentrations, and bands further along indicating shorter sequences.
PCR
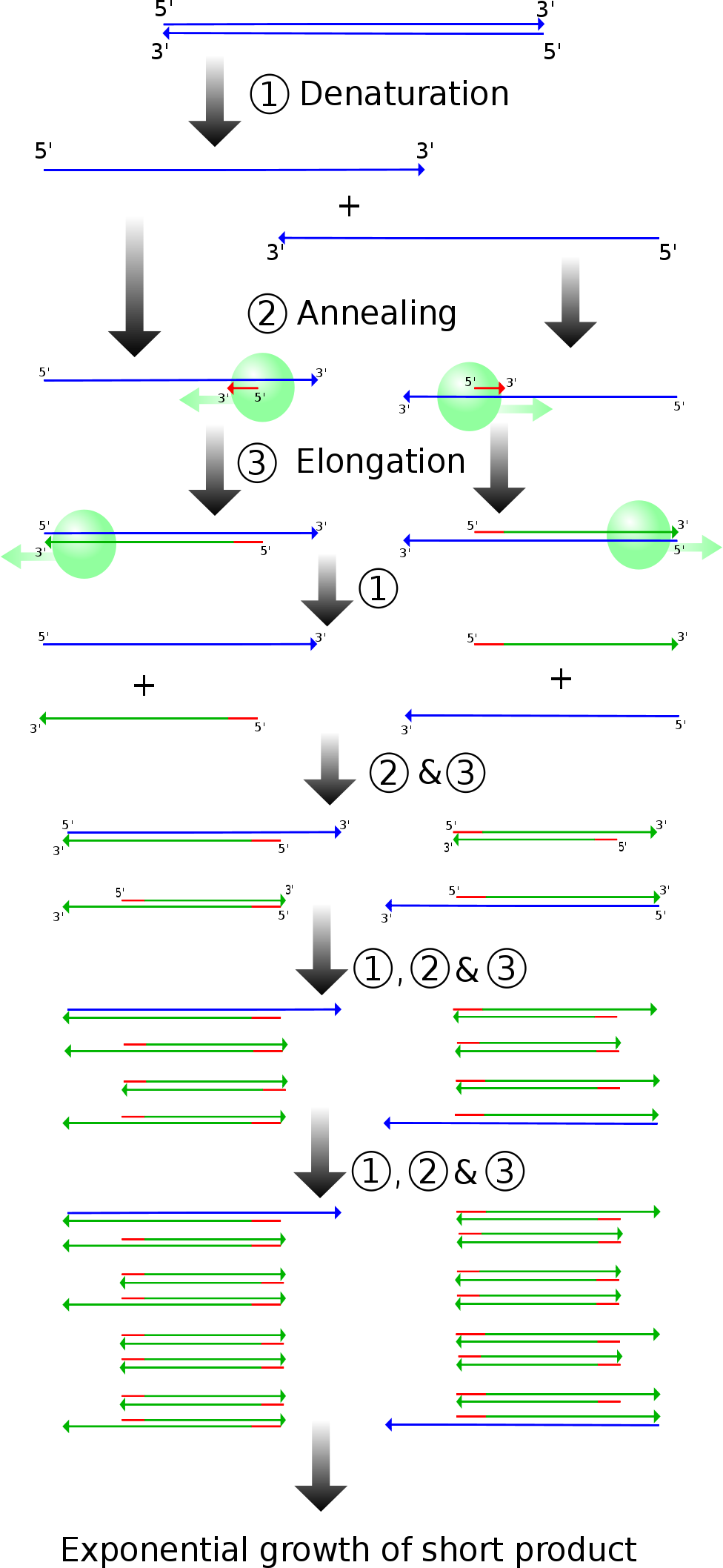
(Madeleine Price Ball, Creative Commons License, sourced from https://en.wikipedia.org/wiki/File:PCR.svg)
Polymerase Chain Reaction (PCR) is a very common technique used to amplify specific sequences of DNA. This can be useful for sequencing, cloning, and other techniques that require high concentrations of DNA.
PCR is run in a solution with DNA containing the target sequence you wish to amplify, primers, and DNA polymerase. First, the solution is heated to near boiling (94\(^\circ\)-96\(^\circ\) C), so that the DNA denatures and becomes single stranded. Once the DNA is single stranded, primers can bind to this DNA.
The primers are designed to bracket the target sequence. The primers must be complementary to the 3’ end of both the template and coding sequences. After the DNA denatures, the temperature is lowered to allow the primers to anneal to the DNA strands. Once the primers are bound, the temperature is raised once more to the optimal temperature of the polymerase being used. A common polymerase is known as Taq Polymerase, and has an optimal temperature of approximately 75\(^\circ\) C. The polymerase binds to the primers and synthesizes complementary DNA until it reaches the end of the strands.
Note that in the very first cycle, this creates long pieces of DNA that go “indefinitely” off to one side of the target sequence. However, in future iterations, the primers bind such that the RNA polymerase synthesizes towards the short end of the strand. Thus, only the target sequence gets amplified exponentially. Due to this exponential amplification, PCR is very sensitive, and can be used to amplify sequences with very low initial concentrations. At 100% efficiency (which is not entirely realistic), the concentration of target sequence would double every iteration, and there are usually somewhere between twenty and thirty iterations of PCR.
Quantitative PCR (qPCR)
Quantitative PCR (qPCR) is an application of PCR in which instead of amplifying the target sequence, you instead wish to measure the concentration or relative concentration of the target sequence in the initial sample.
PCR begins and proceeds as in a usual reaction. However, the concentration of the target sequence is measured after every iteration of PCR. One common way to do this is to have a double-stranded DNA binding fluorophore; thus, when the DNA is allowed to cool and return to its double-stranded shape, the dye binds to it and exhibits fluorescence. The more DNA is present, the more dye binds and thus the greater the fluorescence. This allows us to measure and quantify the levels of DNA present after each iteration.
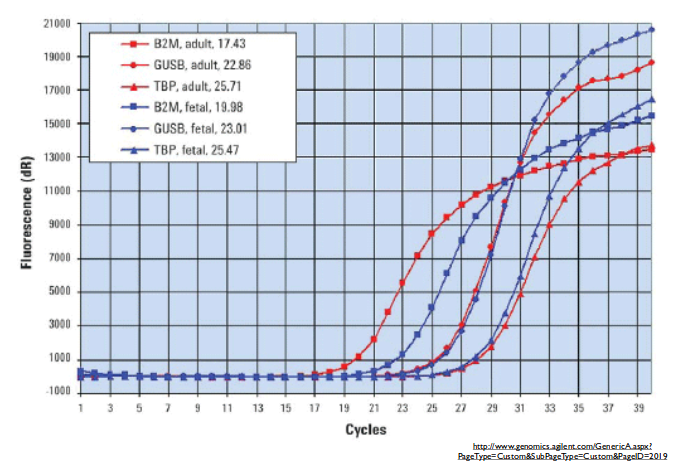
We get concentration plots that look like the plot above. Initially, the reaction runs exponentially, with no limiting conditions. At the end of the reaction, however, it begins to run out of primer or nucleotide components, and thus slows down until eventually it reaches a steady state after which there is no more amplification. Together, these form what is essentially a logistic curve.
In order to extract information from this, we can do one of two things. First of all, we can fit a logistic curve to our data and use that to extrapolate to the start of the reaction in order to determine the relative initial concentrations of the target sequences. We can also just measure the vertical delta after a given number of cycles (close to the beginning of the amplification, while it is still exponential, and not towards the end), and compare those values to determine which has higher amplification. (In the figure above, for instance, what is reported just corresponds to amplification after a given number of cycles.)
Reverse Transcription
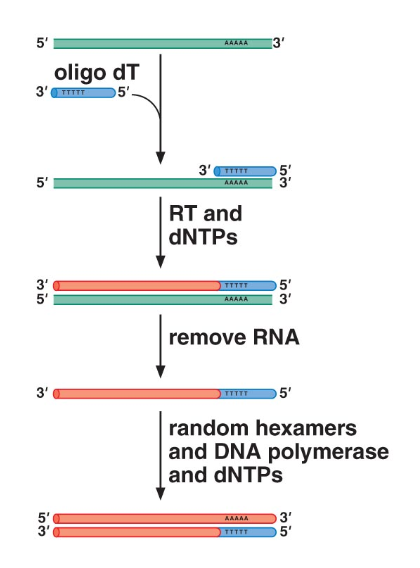
Reverse transcription is a process by which we can convert strands of RNA back into DNA. The generated DNA is known as complementary DNA (cDNA), and is created by enzymes known as reverse transcriptases.
The process is initiated with an oligo (which acts as a primer), reverse transcriptase, and nucleotides (dNTPs). The transcriptase synthesizes a complementary DNA strand to the given RNA sequences. Then, the RNA is removed, leaving a DNA strand with the RNA primer at the beginning. Afterwards, DNA polymerase synthesizes a strand complementary to that one entirely out of DNA, yielding a copy of the original RNA as a DNA strand.
Note that due to the complexity of this process (multiply involved polymerases, etc), it turns out to be incredibly error prone and causes a rather high mutation rate. Retroviruses, for example, have a genome that consists entirely of RNA, and use reverse transcription to reproduce; this results in a very high mutation rate among them.
DNASel Footprinting
In class, we discussed the lac operon, and how the repressor binds to a region next to the DNA that the RNA polymerase covers.
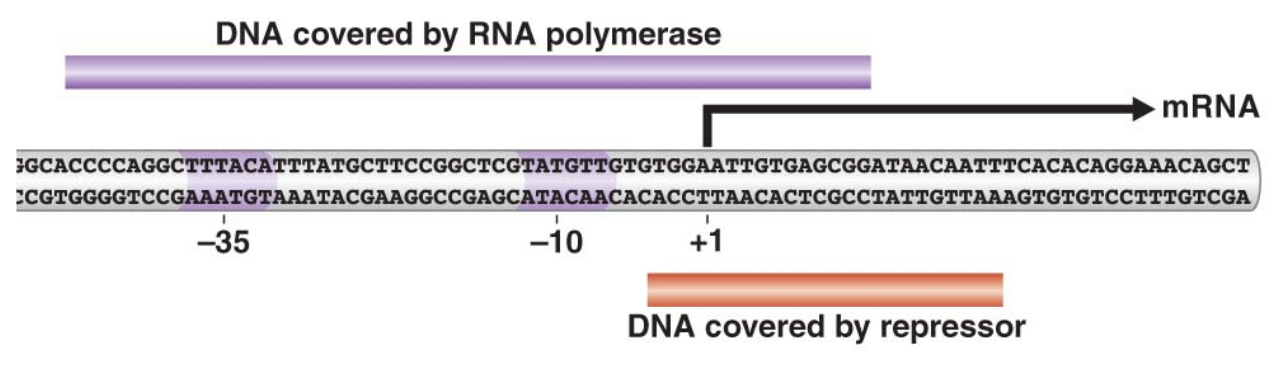
We saw the figure above, which depicts the regions of the DNA that each of the proteins covers when bound to the DNA. This figure leads to a natural question — namely, how can we determine the data presented?
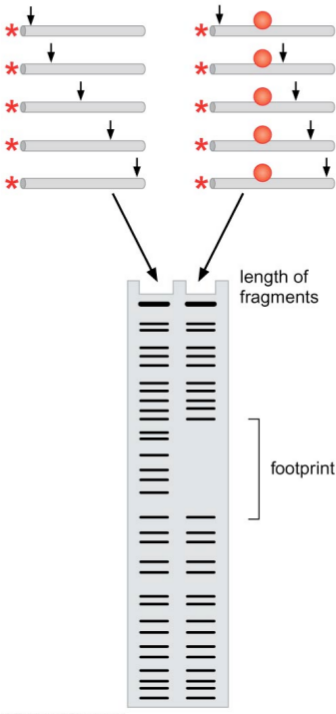
DNAsel footprinting is a technique for determining which bases a protein covers when it binds to the DNA. Note that this does not just determine the points of contact of the protein to the DNA, but instead determines which bases are covered (and thus prevented from binding to other proteins).
DNAsel footprinting is an in vitro technique. Begin by taking some DNA of interest (such as the lac promoter and the region around it) and radioactively labeling the 5’ end of the DNA (with \(^{32}\)P). Next, treat the DNA with DNAse1. DNAse1 is an endonuclease. While exonucleases is something which cleaves nucleotides from the end of a polynucleotide, an endonuclease cleaves polynucleotides starting at some position in the middle and going from 5’ to 3’. DNAse1 is an endonuclease which has no affinity for a particular DNA sequence, so it effectively cleaves the DNA at a random position.
Once the DNA has been treated with the endonuclease, use an acrylamide gel to visualize the radioactively labeled sequences. Since we only labeled the 5’ ends of the sequences, only the lengths of the 5’ ends of the DNA fragments will be visualized. Note that we calibrate the amount of DNAse to the concentration of DNA, in order to prevent the same DNA from being cleaved multiple times.
In addition to doing this reaction with the DNA of interest, we also do it with the same DNA in the presence of the protein of interest. The protein will bind to the DNA, and cover some of the bases. Then, when the DNAse attempts to bind to the DNA at those bases in order to cleave the DNA, it will be unable to bind. Thus, there will be a gap in the fragment lengths — a footprint — in the locations where the endonuclease was unable to bind. If we run a simultaneous Sanger sequencing reaction, we will know exactly where that gap was and what bases it corresponds to, and this will tell us which bases the protein covers.
Note that this procedure isn’t completely precise. For instance, the endonuclease itself takes up some number of bases, so there may be a part of the DNA strand that is never cleaved but is also uncovered by the protein we’re interested in — it is simply close to the covered bases. This makes DNAsel footprinting slightly overestimate the size of the covered region, but usually by no more than (approximately) ten nucleotides.
Yeast two-hybrid assay
In class, we discussed the following figure, in which we can see RNA polymerase binding to a promoter as well as the \(\alpha\)-CTD subunit binding to the activator CRP.
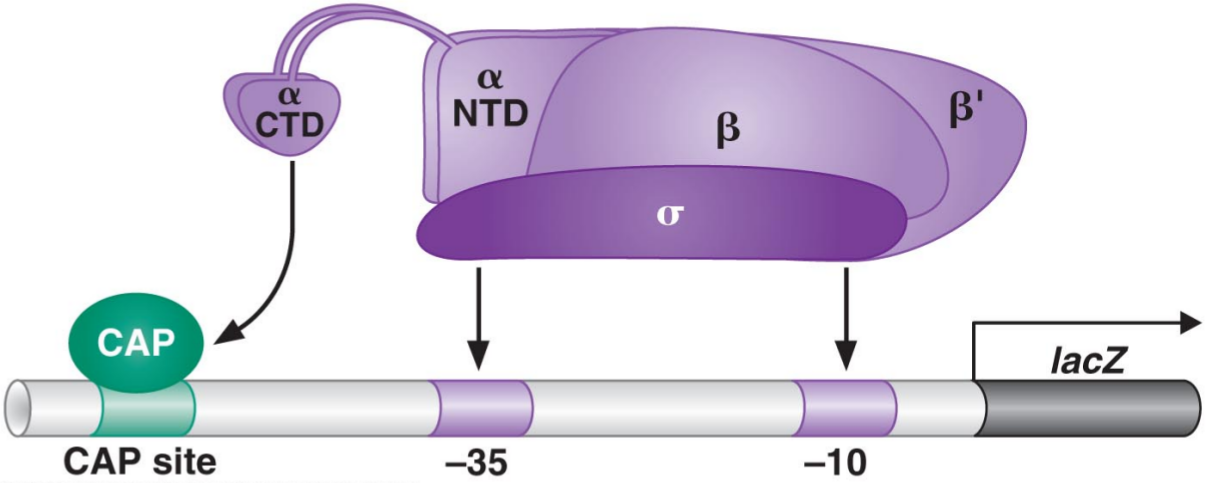
This led to a natural question: suppose that we didn’t know about CRP but instead wanted to find all proteins that bound to a given target protein. How could we do this?
The yeast two-hybrid assay is an approach to doing this. It relies on the fact that activators, such as the Gal4 activator (which binds to a Gal4 binding site and is involved in galactose metabolism in yeast), are composed of two functionally separate pieces. One of them contains the DNA-binding domain (which has the function of chemically bonding with the DNA in the binding site), while the other contains the activating region, which is responsible for binding to the RNA polymerase in order to increase its affinity and thus activate transcription.
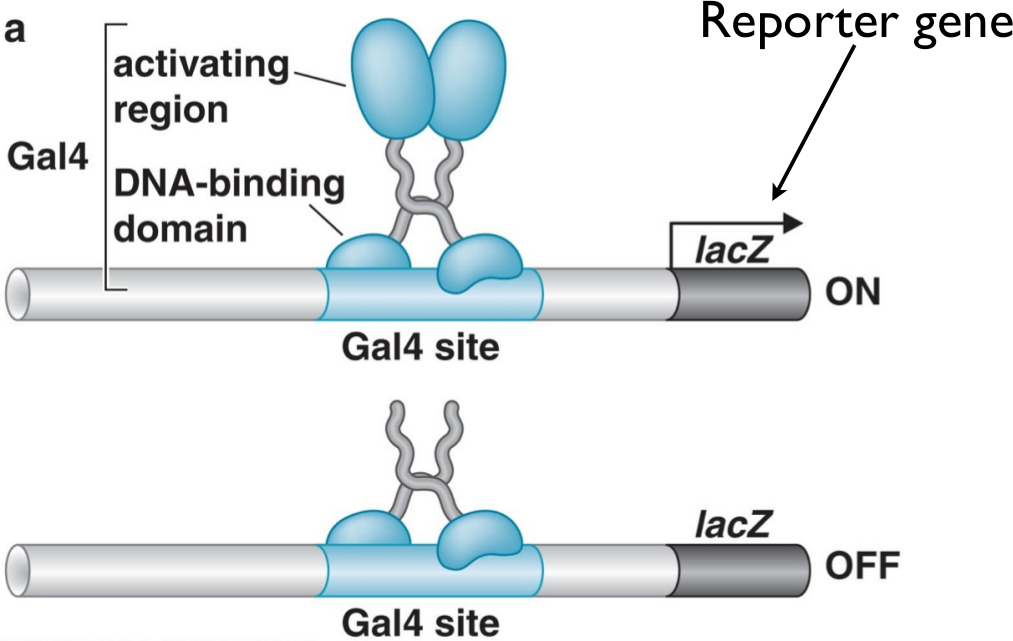
The key insight is that we can separate these two regions in the DNA itself. Using restriction enzymes and ligation techniques we’ve discussed previously, we can separate the DNA binding domain and the activating region into two separate plasmids. Then, we can fuse our protein of interest (via these same techniques) into the DNA binding domain. The resulting complex can be referred to as the bait, because it will bind only to proteins that interact with our target protein.
We then generate a very large library of proteins which are attached to the activation domain sequence. In fact, each plasmid we generate may have a separate protein sequence attached to the activation domain. Once we have bait and prey plasmids, we can force yeast to incorporate these plasmids.
Suppose that a yeast cell incorporates the bait plasmid and a prey plasmid which happens to have the sequence for the activation domain bound to a sequence for some protein that the protein of interest interacts with. Then, when the bait and prey are translated, the DNA binding domain (attached to the bait) will bind to the DNA next to the LacZ promoter, while the prey will interact with the bait, thus bringing the activator domain in proximity to the LacZ promoter. As a result of this interaction, the LacZ gene will be activated and highly expressed in the cell.
Suppose, instead, that a yeast cell incorporates a prey plasmid which has a protein coding sequence for a protein that does not interact with the bait. In that case, the activation domain will never be brought close to the promoter, and thus the LacZ gene will not be expressed nearly as strongly as it is in the other case. Thus, expression of LacZ is tightly coupled with whether a yeast cell incorporated a prey plasmid for a protein that interacts with the target protein.

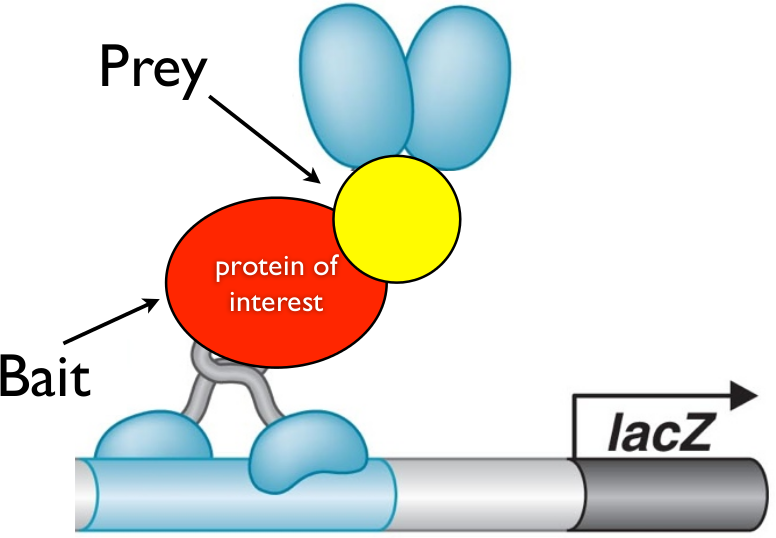
(Bottom) When prey protein interacts with bait, activation domain is proximal to the promoter, and thus transcription is activated.
Yeast cells on a plate will form distinct colonies, each of which is descended from a single cell, so each colony will have the same plasmids that the mother yeast cell contained. We can then use X-gal to test for the expression of LacZ. When the LacZ gene is expressed, it creates mRNA which codes for a protein known as \(\beta\)-galactosidase. \(\beta\)-galactosidase cleaves X-gal (which is composed of a particular molecule bound to a galactose) into the constituent parts; while X-gal itself is colorless, the X molecule alone (without the galactose) is visibly blue. Thus, in the presence of X-gal, the yeast cells which have prey that interacts with the bait and thus have strong expression of LacZ will be visibly blue, while the others will be colorless.
We can use the color of yeast colonies to separate out the colonies which received prey plasmids that interact with the bait. We can purify out the prey plasmids from these blue yeast colonies and sequence them, yielding the sequence of a protein that interacts with our target protein (which was attached to the bait protein).
Although this technique can be very useful, it has a number of problems as a result of which it is a much better hypothesis generating tool (as opposed to one which can be used to check a hypothesis). This technique can easily fail to find proteins that interact with our target protein, because it may be that the proteins need to be modified post transcriptionally (which may not happen when they are bound to the activation domain) or perhaps they must be localized to a particular part of the cell. Similarly, the technique can find proteins that do not actually interact with the target protein — for instance, if the target protein is only expressed in high temperature environments, the library may include a protein that seems to interact with the target protein but actually has no biological relevance, because it is only expressed in low temperature environments (and thus the interaction can never actually happen). The technique may fail to find proteins simply because, due to the complexities of protein folding and interactions, glueing together the sequences of two proteins will not necessarily yield a protein with the same interactions as the separate proteins.
Western Blotting
In previous technique primers, we discussed Northern and Southern blotting, which are techniques used to visualize the abundance and potentially sizes of RNA and DNA, respectively. A similar technique exists for proteins and is appropriately named Western blotting, or occasionally Western hybridization.
The fundamental difference between Western blotting and Northern and Southern blotting is the probe. While RNA and DNA can both use a similar RNA probe, we cannot use an RNA probe to visualize proteins. Instead, we must use antibodies.
Antibodies are large Y-shaped proteins produced by immune systems to identify and destroy foreign cells and viruses. For the purposes of Western blotting, the protein we’re interested in targeting may be introduced to a cell culture or a mammal (such as a mouse) in order to induce an immune response. The immune system will produce an antibody (known as the primary antibody) which binds to the target protein. We can purify this antibody from the cells and then use it as a probe.
Once we have an antibody probe, the process of a Western blot is very similar to that of a Northern or Southern blot. We run our proteins through a gel, which separates the proteins based on their size; we transfer these proteins to a membrane; we block any unused area on the membrane to prevent antibodies from binding there by placing the membrane in a solution of non-specifically binding protein; finally, we visualize the results by letting the labeled probe antibody bind to the target proteins.
Note that although it is possible to label the primary antibodies (those created by the immune system response) with fluorescent molecules and then visualize them, this can be rather expensive, as each antibody must be custom. However, instead, we can use a fluorophore-labeled secondary antibody. Secondary antibodies are antibodies which bind to the primary antibodies; however, unlike primary antibodies, secondary antibodies will bind to almost any primary antibody, and are thus nonspecific and much cheaper.
Chromatin immunoprecipitation (ChIP)
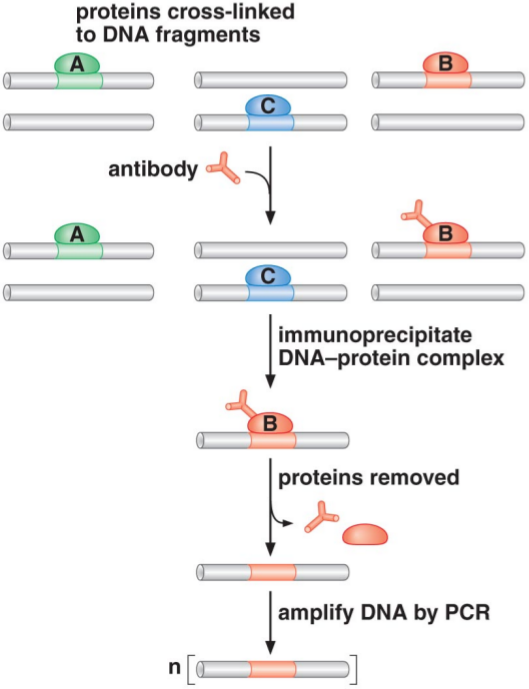
While the yeast two-hybrid assay can be used to detect all proteins that interact with a particular protein, we may also be interested in detecting all DNA locations which a given protein interacts with. We can use chromatin immunoprecipitation, a technique which precipitates out DNA sequences which interact with a protein, in order to detect these DNA locations.
When doing chromatin immunoprecipitation, cells are first fixed with a cross-linking agent, such as formaldehyde or ultraviolet light. Cross-linking is a reaction in which a protein can be covalently bound to DNA. At this stage, all proteins may be bound to DNA in various places, as we can see in the first row in the image to the right. We then shear DNA, which breaks it into fragments only several hundred bases in length. (If we did not shear the DNA, we would end up precipitating the entire chromosome, instead of only the regions which had proteins bound to them.)
Next, we mix this solution with antibodies specific to the protein we wish to target. These antibodies are created in a manner similar to the one we used for Western blots. The antibodies bind to the protein we are interested in (which is still cross-linked to the DNA regions it interacts with). We can then couple these antibodies with another molecule, such as agarose or magnetic beads. These molecules form the immunoprecipitate, which allow us to precipitate out or otherwise separate the antibodies and everything they’re bound from the rest of the solution.
Once we have separated these out, we remove all the bound proteins. (This is why we require that the cross-linking be reversible.) Once we remove the proteins, we are left with only DNA. We can then amplify this DNA through a polymerase chain reaction (PCR). Sequencing this DNA will give only the fragments which contain the DNA sequences which the target protein binds to, thus identifying those sequences.
Electrophoretic mobility shift assay (EMSA/gel shift)
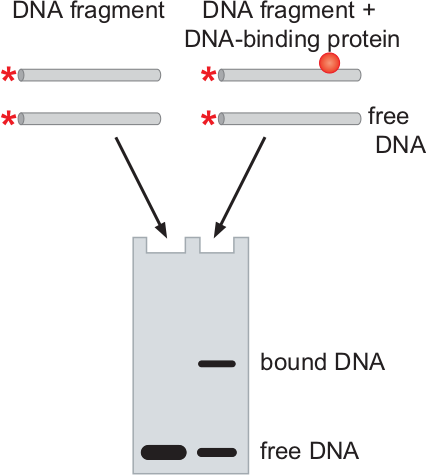
In addition to locating which DNA a protein interacts with, as we can do with chromatin immunoprecipitation, we may want to determine whether a protein interacts with a piece of DNA, or the order in which several proteins interact with a DNA binding site. This type of experiment may be done via an electrophoretic mobility shift assay (EMSA), commonly known as a gel or band shift assay, and can be very useful for studying processes such as transcription (especially initiation) and DNA replication.
The fundamental fact that EMSA relies upon is that DNA strands that have proteins bound to them will move slower in an electrophoresis gel than DNA strands with no proteins bound to them, and that larger bound proteins will result in a slower movement (lower electrophoretic mobility) than smaller bound proteins.
We begin by taking a single or double stranded piece of DNA containing the binding site of interest and labeling it radioactively. Although we can label it with a fluorophore, this may be harder, because the fluorophore must be placed such that it does not interfere with the binding of proteins to the DNA. This labeled piece of DNA is sometimes referred to as the “probe”.
Next, we mix our probe with the protein (or proteins) of interest. We let them interact and then separate out the molecules of different electrophoretic mobilities in a nondenaturing gel. Note that unlike other techniques we’ve discussed, the gel must not denature the molecules, as that would cause them to unbind and render the entire technique pointless. In another lane of the same gel, we can run the DNA probe without the proteins. We can then image the gel and observe the bands in both gels; if the protein binds to the DNA, then we will note a significant shift in the band. The diagram on the left, for instance, depicts a single protein binding to the DNA; you can see that some of the DNA was bound, producing a band closer to the start location (since it moved slower), while some of the DNA remained free, thus creating a band in the same place as the unbound DNA on the DNA-only lane.
Note that we can use EMSA to study the interactions of multiple proteins as well. These interactions can be due to multiple binding sites, or even due to subsequent proteins interacting with proteins that have bound to the DNA. As each additional protein binds, it will reduce the mobility, thus causing a greater shift. This can help identify how a series of proteins interacts with a strand of DNA — since larger proteins will slow the DNA down more than smaller once, you can distinguish which protein bound to the DNA. (When you have two identically sized proteins, you can differentiate between them by adding an antibody that targets only one of them; the antibody will bind to that protein, effectively increasing its size and allowing you to differentiate from the other one. This technique is known as a supershift assay.)
One weakness of this technique is that while it can confirm binding of protein to DNA, it cannot identify the precise binding site in the DNA strand. However, other techniques may be used to rectify this. For instance, mutations may be introduced into the DNA strand to assess the specificity of the protein binding. Another common technique is to run extra lanes with different oligonucleotides which compete with the DNA strand for binding; if you use oligonucleotides of known sequence, you can identify the sequence of the binding site in the DNA.